本文刊于:中华儿科杂志, 2018,56(3) : 179-185
作者:于仲勋 钟林庆 宋红梅 王长燕 王薇 李冀 马明圣
单位:中国医学科学院 北京协和医学院 北京协和医院儿科
摘要
目的
总结中国首例干扰素基因刺激蛋白(STING)相关婴儿期起病的血管病(SAVI)的病例特点和初步疗效观察。
方法
分析2017年11月北京协和医院儿科收治的1例高度疑似SAVI患儿的病例特点,并用Sanger测序进行患儿及其生物学父母相应突变位点验证明确诊断。以"STING""SAVI""自身炎症性疾病""干扰素通路疾病"为检索词,检索2010年1月至2017年12月中文数据库及Embase、Pubmed数据库进行文献复习。
结果
患儿 男,14岁,生后起病,以反复干咳伴活动耐力下降为主要表现,伴有生长受限、反复冻疮样皮疹、毛细血管扩张和长的杵状指,实验室检查:红细胞沉降率78 mm/1 h,C反应蛋白21 mg/L,免疫球蛋白G 22.16 g/L,胸部高分辨CT提示间质性肺病,超声心动提示肺动脉高压[61 mmHg(1 mmHg=0.133 kPa)]。经Sanger测序发现患儿TMEM173基因新生杂合突变:c.463G>A, p.V155M,经托法替尼5 mg,2次/d治疗后患儿活动耐力较前增加。文献检索共8篇,中文0篇,英文8篇,全世界目前共有20例SAVI病例,多伴有生长受限、皮肤和肺部的受累。
结论
SAVI是自身炎症性疾病中干扰素通路疾病中的一种,多以反复冻疮样皮疹、间质性肺病、生长受限、免疫球蛋白G升高、炎性指标升高为临床表现,Janus激酶抑制剂对该病有一定疗效。
自身炎症性疾病是一组包括家族性地中海热、肿瘤坏死因子(TNF-α)受体相关周期热综合征、高IgD综合征等疾病在内的以反复发热和严重炎症反应为特点的疾病。干扰素通路病是自身炎症性疾病领域内的重要成员。2014年,N Engl J Med发表了6例干扰素基因刺激蛋白(STING)相关婴儿期起病的血管病(STING-associated vasculopathy with onset in infancy, SAVI),该6例均由STING编码基因TMEM173杂合新生功能获得性突变所致,其病例特点包括新生儿期全身炎症反应,严重的皮肤血管病导致组织坏死丢失和间质性肺病[1]。现报道中国大陆地区首例SAVI病例的临床表现及基因突变位点,同时为该罕见病的治疗提供初步诊疗经验。
临床资料
一、本组病例资料
患儿 男,14岁,因"反复咳嗽伴活动耐力下降14年"于2017年11月入院。患儿自2月龄起大笑及哭闹后反复干咳,安静后可缓解,不伴发热,无明显口唇发绀等表现。8月龄练习爬行,家长发现不愿动。1岁学会走路后不愿主动行走。患儿自幼活动后时常表达"抱"的意愿,可见患儿呼吸频率增快,可伴有干咳。3岁后与同龄儿玩耍时不愿奔跑、打闹,诉追赶不过同龄儿。6岁时因受寒后发热、干咳、活动后气促,就诊于当地医院,查胸X线片示肺炎、CT可见肺部"小囊状改变",抗感染治疗后体温恢复正常,但干咳无好转,予"抗哮喘"治疗后咳嗽亦无好转。此后每年1~3次"肺炎",症状同前。8岁后双耳反复冻疮,偶有口唇及四肢发绀、面色苍白。9岁时因身高增长较缓慢予生长激素治疗3个月,治疗后身高增长6~8 cm。10岁时因阑尾炎行腹腔镜阑尾切除术,同年出现脱发,表现为斑秃,持续半年后自行缓解。12岁时因军训再次出现咳嗽伴呼吸困难,就诊于外院,诊断为间质性肺病、轻度肺动脉高压[超声心动图测肺动脉压力71 mmHg(1 mmHg=0.133 kPa)],予甲泼尼龙2 mg/kg治疗后无显效且进展为重度肺动脉高压(超声心动图测肺动脉压力107 mmHg),遂停药并于外院行右心导管术及双侧肺动脉扩张治疗,提示毛细血管前肺动脉高压。既往及个人史:1月龄出现双侧面颊部及颈前淡红色皮疹,不突出皮面,局部按压可稍褪色,哭闹后颜色稍加深,偶可见毛细血管扩张,外用激素类药物皮损可减轻,皮疹9月龄后逐渐消退。患儿系第1胎第1产足月经顺产娩出,出生体重3.3 kg,身长50 cm,头围34 cm。生后无窒息,吃奶好。母亲孕4个月可疑弓形虫感染,生后2月龄抬头,6月龄会坐,按计划疫苗接种。
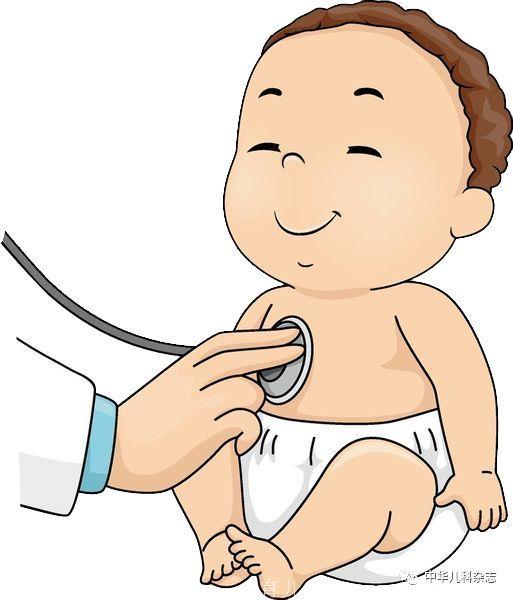
入院体检:身高155 cm(第3~10百分位),体重30 kg(小于第3百分位),体温36.2 ℃,心率90次/min,呼吸频率26次/min,血压105/67 mmHg,经皮血氧饱和度0.04(室内不吸氧)。营养欠佳,皮下脂肪少。枕部及背部皮肤可见散在扩张的毛细血管(图1A、图1B),右耳廓冻疮样皮疹(图1C),全身浅表淋巴结未及,头颅大小正常无畸形和压痛,头颅毛发无异常。颈静脉无怒张,双肺呼吸音稍粗,可闻及明显爆裂音,未闻及湿啰音及胸膜摩擦音,心前区无隆起及凹陷,心界基本正常,心率90~100次/min,心音有力,心律齐,P2>A2,三尖瓣听诊区可及2/6级收缩期吹风样杂音。周围血管征阴性。腹软,无压痛、反跳痛,肝脾未及,Murphy征阴性,肠鸣音正常。脊柱无畸形、压痛,四肢关节活动自如,四肢无浮肿,杵状指(图1D),略长,肢端稍发绀,双足背动脉搏动正常。肌力正常,生理反射存在,病理征阴性。
入院后对患儿进行多系统评估。血常规:白细胞9.25×109/L,淋巴细胞2.81×109/L,单核细胞0.36×109/L,中性粒细胞5.77×109/L,血小板385×109/L,血红蛋白113 g/L;粪常规、尿常规正常;血电解质正常;丙氨酸转氨酶9 U/L,白蛋白38 g/L,总胆红素6.3 μmol/L,乳酸脱氢酶258 U/L,肌酐40 μmol/L,总胆固醇3.56 mmol/L,游离脂肪酸975 μmol/L,乳酸1.26 mmol/L,肌酸激酶35 U/L,心肌肌钙蛋白阴性,N末端B型钠尿肽原未见异常;铁蛋白57 μg/L;降钙素原<0.05 μg/L;真菌D-葡聚糖(G试验)阴性;胰岛素样生长因子116 μg/L(降低);甲状腺功能正常;骨代谢:β胶原降解产物2.300 μg/L(升高),总25羟维生素D 14.4 μg/L,骨密度低于同年龄儿童2个标准差;凝血正常;尿α1微球蛋白、尿β2微球蛋白未见异常。
免疫球蛋白:IgG 22.16 g/L(升高),IgA 4.73 g/L(升高), IgM 0.72 g/L,补体:C3 1.128 g/L, C4 0.217 g/L,淋巴细胞亚群: B细胞比例0.204,B细胞计数0.574×109/L,自然杀伤细胞(NK)比例0.540,NK细胞计数0.152×109/L, T淋巴细胞比例0.739,T淋巴细胞计数2.078×109/L,辅助T淋巴细胞比例0.282,辅助T淋巴细胞0.793×109/L,细胞毒T淋巴细胞比例0.439,细胞毒T淋巴细胞计数1.234×109/L。抗链球菌溶菌素O 6.0 U/ml,类风湿因子154.5 U/ml;抗核抗体谱三项、抗可溶性核抗原抗体谱、抗磷脂抗体谱、系统性血管炎相关自身抗体谱阴性;炎性指标:白细胞介素-6(IL-6) 22.8 ng/L、TNF-α 19.3 ng/L, C反应蛋白(CRP)21 mg/L,红细胞沉降率78 mm/1 h。
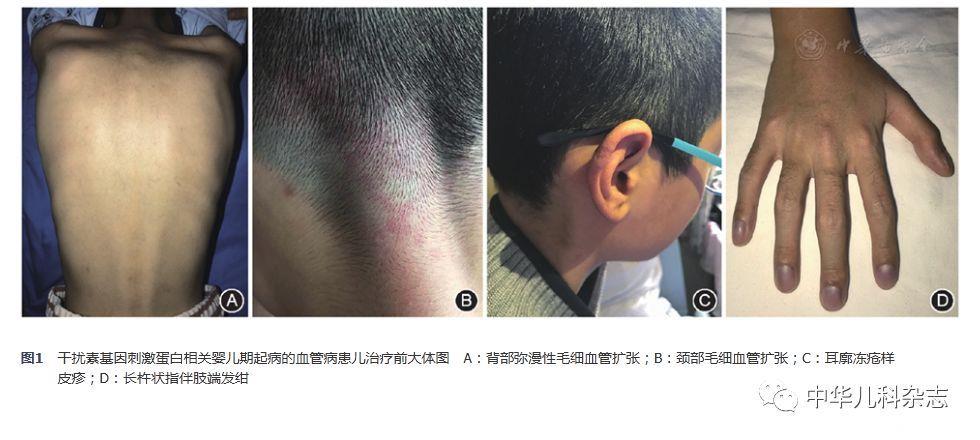
腹部超声:胆囊壁毛糙,肝脾无肿大;肺功能:肺总量占预计值0.457,用力肺活量(FVC)占预计值0.322,第1秒用力呼气容积(FEV1)占预计值0.316,FEV1/FVC 0.816,一氧化碳弥散量占预计值24.1%,提示限制性通气功能障碍伴弥散功能减低;胸部高分辨CT示双肺多发索条、网格、磨玻璃影,部分呈蜂窝状改变,可见囊腔,双下肺为著(图2);我院会诊院外肺活检病理:肺泡腔扩张,其内见大量泡沫状组织细胞聚集,散在少许胆固醇结晶,伴Ⅱ型上皮细胞增生,肺泡间隔增宽,纤维组织增生,免疫组织化学:甲状腺转录因子-1(TTF-1)阳性、CD68阳性、平滑肌肌动蛋白(SMA)阴性、CD34阴性(图3)。超声心动提示右心增大、中度肺动脉高压,左心室射血分数(M型)64%,主肺动脉增宽(24 mm),估测肺动脉压力61 mmHg,三尖瓣环收缩期移位(TAPSE)12 mm,头颅CT及磁共振成像(MRI)未见异常,无基底节区钙化等表现。

根据患儿反复干咳伴发热、冻疮样皮疹、毛细血管扩张、间质性肺病、炎症指标升高的特点,考虑自身炎症性疾病可能性大,SAVI的可能性最大,故经知情告知后留取患儿及其家长的血液标本进行TMEM173的基因测序(引物信息:上游引物5'➝3':GGAACAGTGGGTTTGGCAAT,下游引物5'➝3':GCCTTGGGATTAAAGGATTTGG),结果显示TMEM173基因杂合新生突变(c.463G➝A, p.V155M)(图4)。患儿确诊为SAVI,同时伴有间质性肺病、中度肺动脉高压、右心增大、轻度贫血。

治疗经过及随访:患儿入院后继续予抗肺动脉高压治疗,包括安立生坦、他达拉非的靶向治疗及钙离子通道拮抗剂地尔硫,因骨密度减低补充钙剂(碳酸钙、维生素D),经与患儿家长充分病情告知后,患儿家长自购托法替尼,予5 mg,2次/d试验性用药,服用2 d至该药达到血药浓度后,观察患儿生命体征:体温36 ℃,心率80~90次/min,呼吸频率20次/min,血压100/62 mmHg,经皮血氧饱和度0.96(室内不吸氧),心率较入院有所下降,经皮氧饱和度较前有一定好转,同时患儿食欲有所增加。炎性指标方面,CRP 7 mg/L,红细胞沉降率76 mm/1 h,IL-6 4.7 ng/L、TNF-α 17.4 ng/L,均较入院有一定程度的下降。出院2周后电话随访,患儿一般状况好,平静状态下,不吸氧经皮血氧饱和度0.97~0.98,心率80~85次/min,饮食进餐量增加至治疗前的1.5倍,可坚持打乒乓球等中等强度运动15 min以上。
二、文献复习资料
在PubMed数据库、Embase数据库、万方数据库、中国知网数据库分别以"STING""干扰素通路疾病""STING-associated vasculopathy with onset in infancy"为关键词检索2010年1月至2017年12月文献,检索到16篇文献,中文0篇,英文16篇,排除非临床报道的文献,共8篇[1,2,3,4,5,6,7,8],均为国外报道,共19例,加上本例,目前共有20例SAVI,现将资料完整的17例(含本例)临床资料进行总结(表1)。
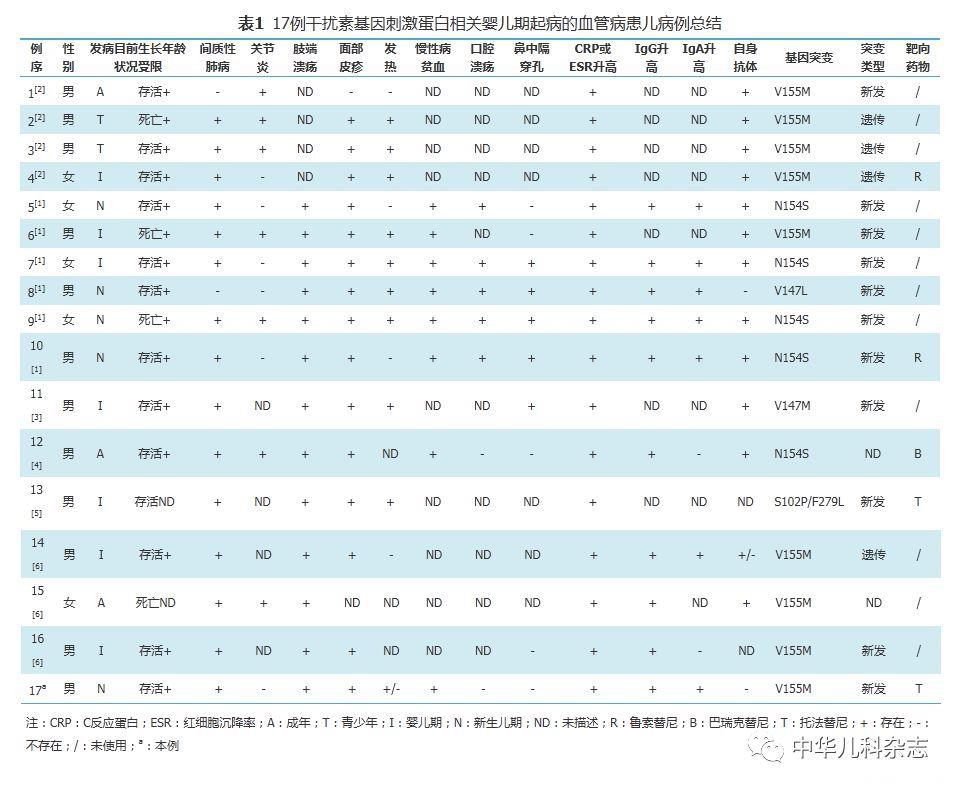
讨论
SAVI是近年来新发现的单基因遗传的自身炎症性疾病[9],也是Ⅰ型干扰素通路疾病家族中的重要成员之一。Ⅰ型干扰素通路病这一概念由Crow[10]在2011年提出,是一组与Ⅰ型干扰素水平异常上调相关的呈孟德尔遗传的疾病。这组疾病多与固有免疫系统的功能失调相关,尽管不同的基因缺陷模式可呈现出不同的临床表型,但所有的疾病均可归因于Ⅰ型干扰素不适当的过度产生,故Ⅰ型干扰素通路病可兼有自身炎症和自身免疫的特点。
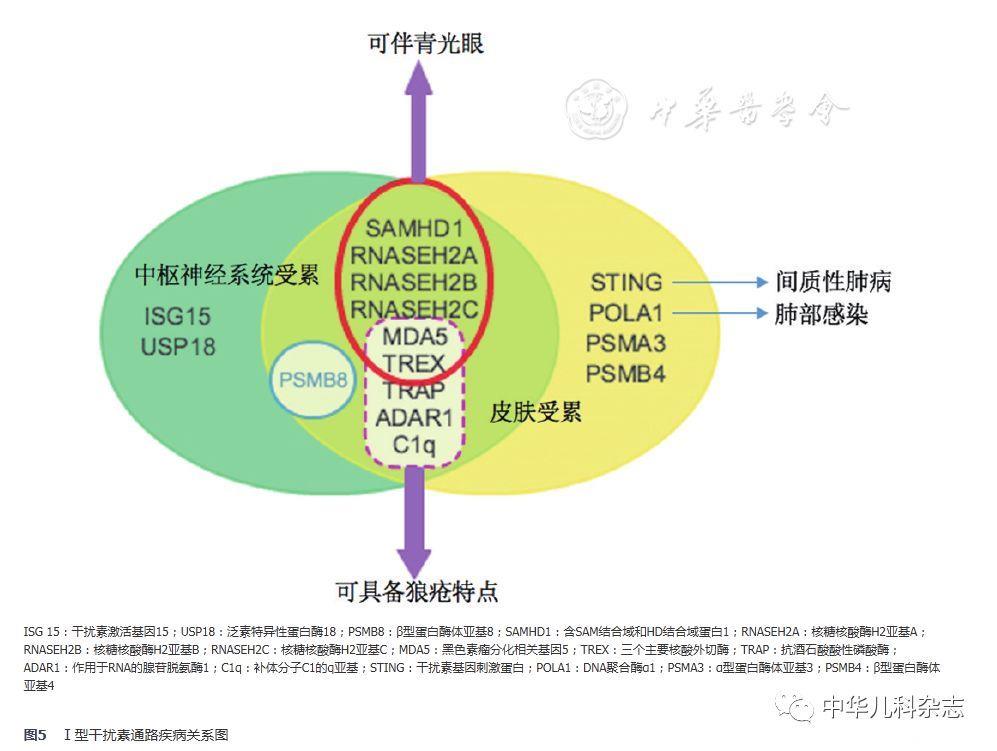
总结目前已发现的干扰素通路疾病,可以发现其过度产生可由以下4种途径引起:(1)内源性核酸的异常集聚或化学修饰;(2)核酸识别过程敏感度的增强或Ⅰ型干扰素信号通路中下游组分的激活;(3)负向调控干扰素信号通路的分子功能障碍;(4)调节Ⅰ型干扰素信号通路的其他通路的缺陷[11]。多种基因突变均可导致Ⅰ型干扰素通路病,目前已知的基因有TREX1、RNASEH2A、RNASEH2B、ADAR1等,以上基因突变可引起Aicardi-Goutières综合征(AGS),其典型表现包括基底节钙化,进行性脑萎缩、小头畸形、肌张力低下等,与胎儿宫内病毒感染引起的症状非常相似[12]。另外,一些干扰素通路的基因突变除了引起典型的自身炎症相关的表现外,还伴有关节炎、自身免疫性肝炎、血小板减少、抗核抗体等自身抗体的阳性和寒冷诱发的冻疮样皮疹等。但总体而言,中枢神经系统和皮肤是干扰素通路疾病最常累及的部位,且干扰素刺激基因的高表达是所有干扰素通路疾病的共同特点。有关干扰素通路疾病的简要总结见图5。
SAVI相关的基因突变位于第5号染色体上的TMEM173基因,目前发现的致病突变位点有p.S102P、p.V147L、p.V147M、p.N154S、p.V155M、p.C206Y、p.F279L、p.R281Q、p.R284G[1,2,3,4,5],其中p.V155M为该基因的热点突变。目前全世界共计报道SAVI20例(含本例),总结显示可散发,亦可在家系中呈常染色体显性遗传模式。对于该病,皮疹为最常见的首发症状,可累及颊部、鼻尖、耳廓、肢端等部位,可伴有皮肤的破损和水泡,若破损继续加重,则可导致肢端的坏死、鼻中隔穿孔等。绝大多数的患者在病程中会出现间质性肺病,并可继发肺动脉高压等并发症。另外,生长受限、慢性病贫血等也是全身炎症反应的间接表现。实验室检查特点包括红细胞沉降率、CRP等炎症指标升高,IgG升高以及自身抗体阳性。本例患儿的临床表现与国外报道类似,起病年龄早,具有典型的毛细血管扩张、冻疮样皮疹、严重的肺间质病变及继发肺动脉高压,红细胞沉降率、CRP、IgG升高。根据该患儿典型的临床表现,且TMEM173基因绝大多数致病突变均位于3号外显子(热点突变p.V155M亦处于3号外显子),因此优先对TMEM173的3号外显子行Sanger测序,结果证实患儿为SAVI。STING是Ⅰ型干扰素信号通路中的转换蛋白。细胞质中的免疫刺激分子如双链DNA在核酸转移酶(cyclic GMP-AMP synthase,cGAS)的作用下产生第二信使环化二核苷酸(cyclic GMP-AMP,cGAMP),STING感知cGAMP后,经由TANK结合激酶1(TANK-binding kinase 1,TBK1)和转录因子干扰素调节因子3(interferon regulatory factor 3,IRF-3)诱导干扰素基因的表达,其模式图见图6。
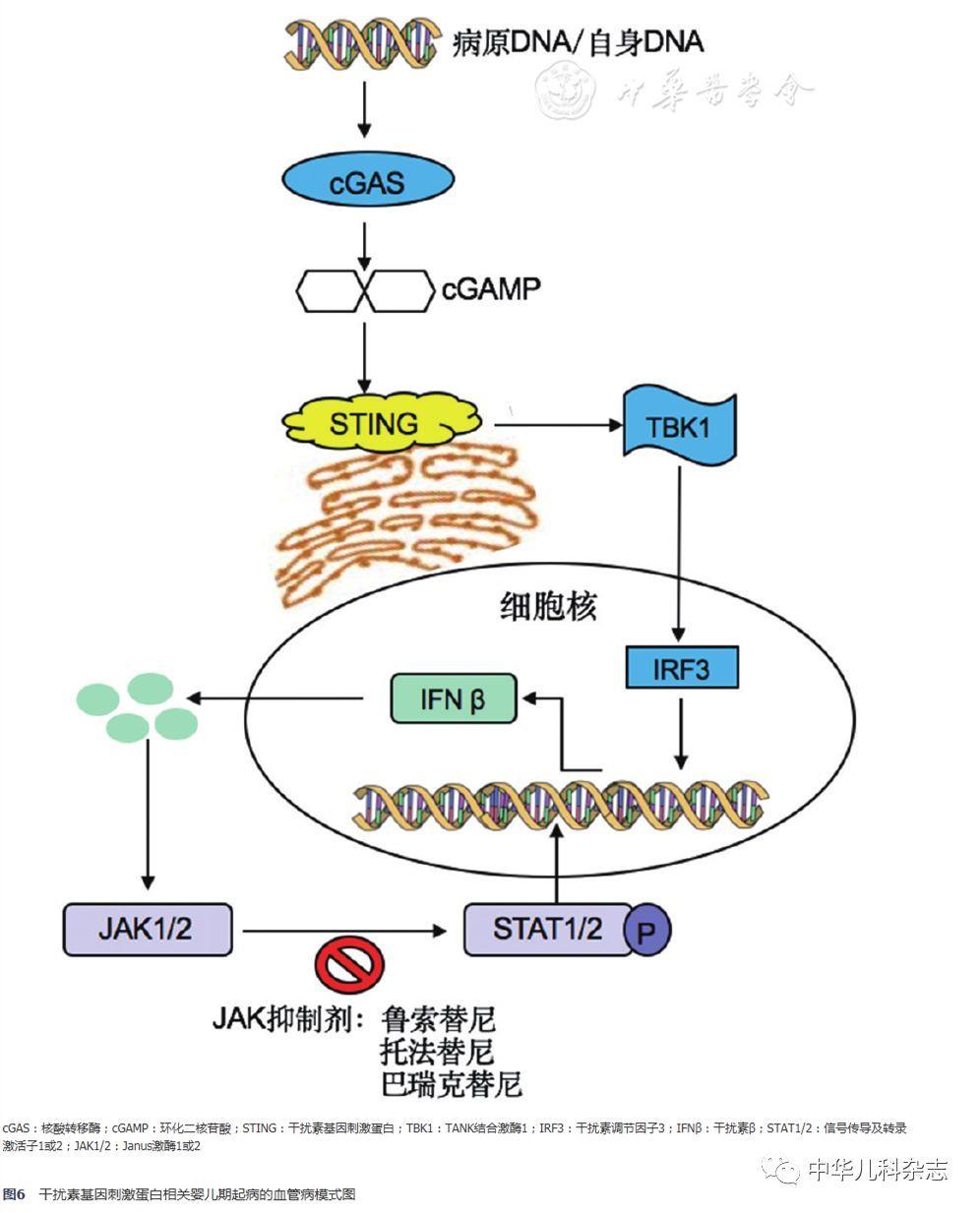
STING广泛表达于内皮细胞、肺泡上皮细胞、巨噬细胞、支气管上皮、自然杀伤细胞、T淋巴细胞和髓系细胞中。STING的过度激活可产生大量的β-干扰素,而β-干扰素与干扰素受体结合后经过JAK-STAT信号通路进一步刺激包括STING在内的高表达,如此循环往复,即可产生大量的炎症风暴,既能引起发热、贫血、关节炎等这些全身表现,又会对STING高表达的组织造成持久破坏。本例患儿炎症指标如CRP、红细胞沉降率均有升高,促炎细胞因子如IL-6和TNF-α的水平亦有升高,提示患儿体内炎症反应不断进行。实际上,根据SAVI发病机制,测定β-干扰素有重要意义,但因p.V155M突变为热点突变,国外高水平文献已进行过极为详尽的功能学实验研究,证实该突变位点的致病性,故本例未再重复进行功能实验。检测到该突变结合患儿典型的临床表型即可明确诊断。而且,患儿存在重度肺动脉高压(曾达106 mmHg),及早地治疗有望改善预后,干扰素测定需要摸索条件,为了不延误患儿的治疗,未进行β-干扰素测定即开始靶向治疗。
SAVI的治疗手段较为有限,大剂量激素和免疫抑制剂无效,已报道的尝试使用的免疫抑制剂包括羟氯喹、硫唑嘌呤、来氟米特、甲氨蝶呤、环孢素A、环磷酰胺、秋水仙碱、沙利度胺均无效。但羟氯喹在体外被证实具有抑制JAK-STAT通路的作用[13]。另外,部分病例尝试使用丙种球蛋白、利妥昔单抗、妥珠单抗、益赛普、英夫利昔单抗亦无明显疗效。因为JAK-STAT通路在疾病的发生发展中也是重要的一环,JAK抑制剂有望成为控制SAVI的良药。已有使用JAK抑制剂控制SAVI的案例报道[4]。JAK抑制剂目前有鲁索替尼、托法替尼和巴瑞克替尼。该患儿目前已使用托法替尼治疗,在服药2 d后CRP水平便降至正常,IL-6水平也降至正常,由此可见,从发病机制入手才能真正实现包括SAVI在内的干扰素通路疾病的控制。
综上,本文报道了中国大陆地区首例SAVI的临床表型和初步疗效观察,同时对全世界范围内所有SAVI病例进行了总结。该病常累及皮肤,以皮肤毛细血管扩张、冻疮样皮疹、肢端溃疡等表现为主,亦累及肺脏,常出现间质性肺病,其他可伴有免疫球蛋白水平的异常改变的自身抗体阳性等。临床医师应对此类自身炎症性疾病加以认识,在接诊反复冻疮样皮疹、活动耐力下降、反复干咳伴炎性指标升高的患儿时应想到本病的可能。
参考文献(略)
欢迎分享到朋友圈,如需转载,请注明转载自《中华儿科杂志》
Strategy
﹀
﹀
﹀
或
是时候和她说再见了!



-Dr.jpg)

