本文刊于:中华儿科杂志, 2018,56(3) : 186-191
作者:余刚 王文婕 刘丹如 陶志锋 惠晓莹 侯佳 孙金峤 王晓川
单位:复旦大学附属儿科医院临床免疫科
摘要
目的
总结人重组激活基因1(RAG1)突变所致免疫缺陷病的临床特征,探讨不同基因突变类型和临床、免疫表型间的关系。
方法
回顾性病例总结。收集2013年10月至2017年6月,在复旦大学附属儿科医院临床免疫科就诊,经临床、免疫评估及基因分析确诊为RAG1基因突变患儿的临床资料,总结临床特征、免疫表型及基因分析结果。
结果
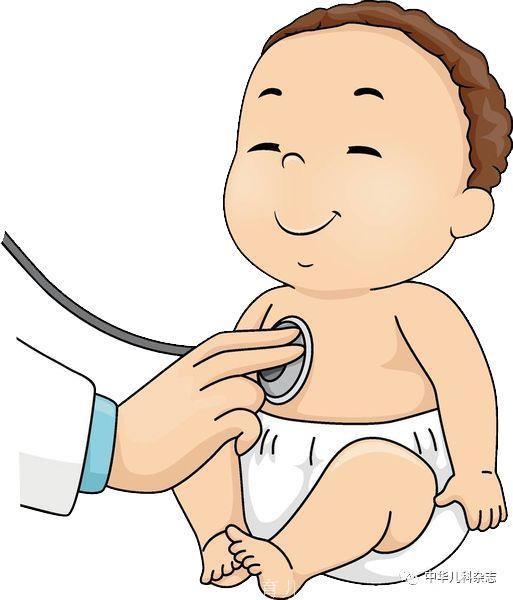
8例患儿确诊为RAG1基因突变,男6例,女2例,起病年龄2~4月龄,诊断年龄2月龄~13岁。4例家庭有婴幼儿反复感染夭折的家族史。2例来自于同一个家庭,其父母为近亲婚配。所有病例均有反复感染,呼吸道(8例)和消化道感染(6例)最多见,其他系统感染包括泌尿道(1例)、中枢神经系统(1例)。感染病原包括细菌、病毒、真菌。轮状病毒感染3例;巨细胞病毒感染5例;卡介苗接种后不良反应2例,其中1例淋巴结穿刺液涂片抗酸染色阳性;真菌感染3例。1例发生肺部、腹腔多处结节性占位伴多发骨质破坏。8例外周血淋巴细胞计数(0.1~3.3)×109/L(中位数0.65×109/L),有3例嗜酸性粒细胞计数增高[(0.48~1.69) ×109/L]。临床表型方面:4例为严重联合免疫缺陷病(SCID),2例为泄漏型SCID,1例为Ommen综合征,1例为联合免疫缺陷病。3例血清IgG低于正常范围,3例血清IgM高于正常范围,5例血清IgE增高。5例为RAG1基因纯合突变,3例为RAG1基因复合杂合突变,6例为已报道突变位点,2例为新发突变位点。3例行造血干细胞移植,4例死亡,1例门诊随访。
结论
RAG1基因不同突变方式导致临床表现多样,临床医生应重视家族史,对于存在婴儿期感染夭折的家庭尽早对患儿进行免疫功能评价和基因检测,尽早明确诊断和采取有效治疗。
人重组激活基因(recombinase activating gene,RAG)是免疫球蛋白(immunoglobulin,Ig)和T淋巴细胞受体(T lymphocyte receptor,TCR)基因片段重排所必需,RAG1基因突变使其编码的重组酶活性完全或部分丧失,V(D)J重组失衡,T淋巴细胞和B淋巴细胞的发育在早期被阻断,导致原发性免疫缺陷病(primary immunodeficiency, PID),为常染色体隐性遗传。有研究指出约50%的不表达T淋巴细胞和B淋巴细胞的严重联合免疫缺陷病(T or B severe combined immunodeficiency, SCID)由RAG1和RAG2基因突变引起[1],但不同的RAG1基因突变类型可引起多种临床表现和免疫表型。现报道复旦大学附属儿科医院经基因分析确诊为RAG1基因突变的病例,总结其临床表特征、免疫表型及基因突变类型。
对象和方法
一、对象
选择2013年10月至2017年6月复旦大学附属儿科医院临床免疫科收治的确诊为RAG1基因突变的8例PID患儿为研究对象,本研究通过复旦大学附属儿科医院伦理委员会批准,批准号:复儿伦审[2013]094号。
二、方法
结合文献[2],生后早期发生反复感染的病例进行临床免疫功能初步评价,包括外周血细胞计数、流式细胞术,常规免疫功能提示免疫缺陷者进一步行全外显子测序(whole exome sequencing,WES)等;依据文献[3,4]中SCID定义为CD3+<300/μl,泄露型SCID为2岁以下患儿CD3+<1 000/μl,再结合本研究病例特点,将诊断为RAG1基因突变PID的临床表型分为SCID,泄露型SCID,Omenn综合征(Omenn syndrome,OS)和联合免疫缺陷病(combined immunodeficiency, CID)。
基因检测:经患儿父母知情同意,抽取患儿及父母外周血各2 ml,行全外显子组测序,结果显示先证者存在RAG1基因突变,然后进行一代测序验证,证实父母均存在RAG1基因突变。由明码生物科技公司进行测序。
三、统计学处理
所有临床数据均采用回顾性分析,病例资料采用描述性分析。
结果
一、一般情况
共有8例患儿确诊RAG1基因突变,其中来自河南省2例、安徽省3例、浙江省2例、上海市1例。患儿基本情况见表1。起病年龄2~4月龄。确诊年龄2月龄~13岁。男6例,女2例。例5和8的父亲和母亲为近亲婚配(父亲的祖父与母亲的外祖母为亲兄妹)。例1、3、4、8的哥哥或姐姐婴儿期夭折,均为感染相关,例1姐姐1岁时因"重症肺炎"死亡,余均在1岁以内因"重症肺炎"等感染死亡,但均未有明确是否免疫缺陷病;例5与例8为亲兄弟。所有患儿接种乙肝疫苗和卡介苗。例2和例8均有接种部位反复化脓、结痂,例2有左腋下淋巴结肿大,例8有左腋下、颈部和腹股沟多部位淋巴结肿大。3例(例1、例2和例6)口服脊髓灰质炎糖丸,例6口服脊髓灰质炎糖丸后出现腹泻。
二、临床资料与转归
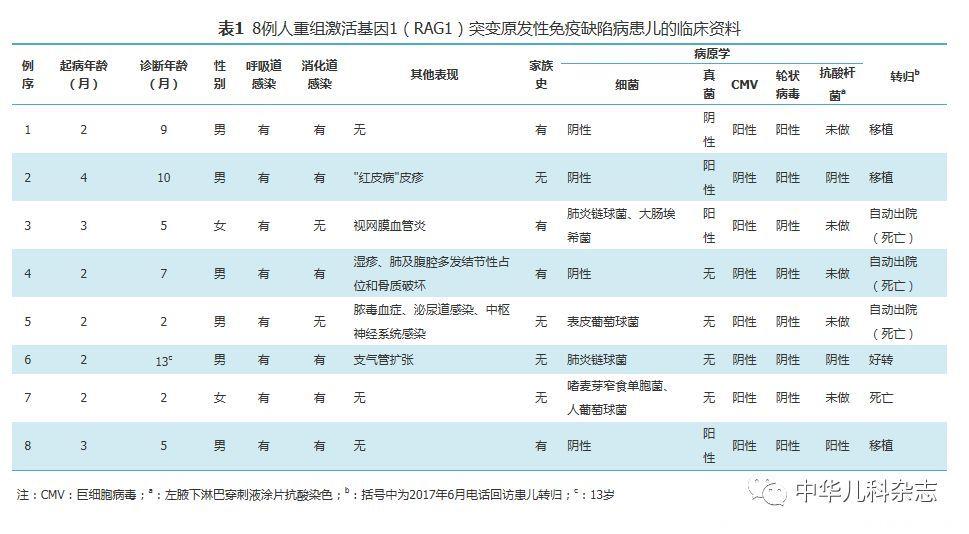
8例患儿的临床资料见表1。8例均有反复或持续呼吸道感染。6例反复腹泻,其中轮状病毒多次阳性3例。例5出现脓毒症、泌尿道感染和中枢神经系统感染。例4影像学发现肺部、腹腔多处结节性占位伴多发骨质破坏(图1)。例2于4月龄起出现反复湿疹伴脱皮,表现为红皮病样,抗过敏治疗控制不佳。肝脾肿大2例(例7、例8)。左腋下淋巴结肿大2例(例2、例8),其中例8淋巴结穿刺抗酸涂片为阳性。呼吸道标本阳性率低,8例中2例(例3、例7)阳性结果,病原包括:肺炎链球菌、大肠埃希菌、嗜麦芽假单胞菌、烟曲霉菌和呼吸道合胞病毒。血培养2例阳性(例5、例7),分别为人葡萄球菌和表皮葡萄球菌。巨细胞病毒(CMV)感染5例(例1、例3、例5、例7和例8)(均为血CMV-DNA滴度增高),其中例3发生视网膜血管炎。有真菌血清学阳性3例,例8血清1,3-β-D-葡聚糖检测(G试验)阳性,例2和例3血清半乳糖甘露醇聚糖抗原检测(GM试验)阳性。
8例患儿4例因感染不治死亡;例6好转出院,现门诊随访中;3例在我院进行造血干细胞移植过程中,均移植成功随访。
三、血常规及免疫学检测结果
8例患儿淋巴细胞计数(0.1~3.3)×109/L,6例淋巴细胞绝对值低于2.0×109/L。中位淋巴细胞计数0.65×109/L。3例(例1、2、8)嗜酸粒细胞计数增高,分别为:1.01×109/L、1.69×109/L、0.48×109/L。例7白细胞减少和粒细胞缺乏,1.69×109/L例8血小板降低,余均在正常范围。例1的CD3+淋巴细胞计数正常,其余均降低;CD4+、CD8+淋巴细胞计数均降低;CD19+淋巴细胞计数均降低;例8的CD16+56+淋巴细胞计数明显升高;CD4+/CD8+升高2例(例2、例5),倒置5例(例1、例3、例4、例6和例8)。例1双阴性T细胞明显增高(68.7%),采用流式细胞仪进一步检测,其中αβDNT为0.07%,γδDNT为58.2%。除例1免疫表型为T+B-外,其余均为T-B-。4例(例1、例3、例6和例8)IgM高于正常范围,其中例6达43.8 g/L(正常值:6.98~14.26 g/L);4例(例4、例6、例7和例8)IgG水平降低,其余病例的IgG升高;IgE水平增高5例(例1、例2、例3、例5和例8),见表2。
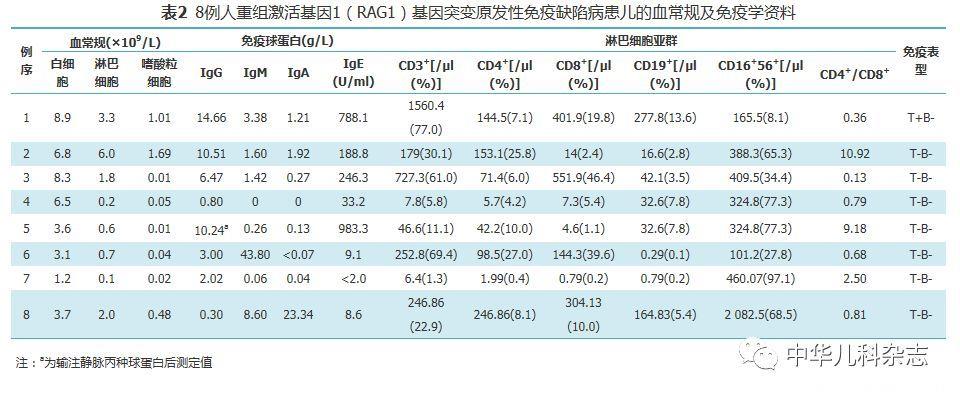
四、RAG1基因突变结果和临床表型
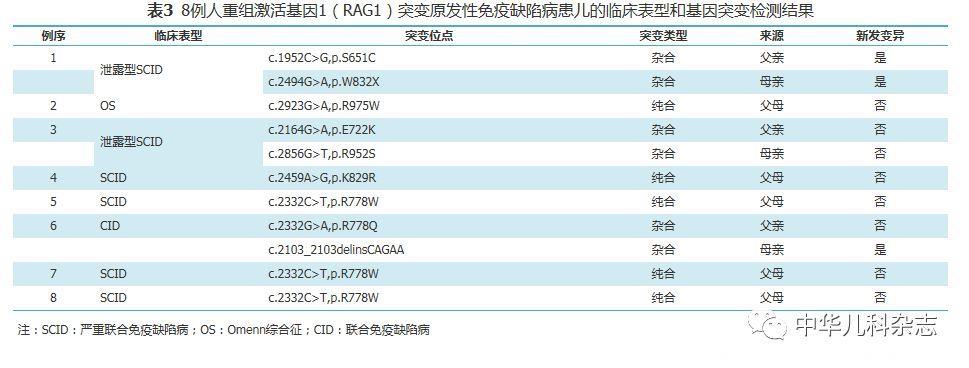
8例患儿有RAG1基因纯合突变和复合杂合突变两种突变方式。5例纯合RAG1基因纯合突变均为已报道突变点位,例4基因突变位点为c.2459A>G,例2为c.2923G>A,例5、例7和例8基因突变位点均为c.2332C>T,例2临床表型为OS,其余4例为SCID。3例(例1、例3和例6)为RAG1基因复合杂合突变,临床表型为泄露型SCID(例1、例3)和CID(例6),见表3。
讨论
RAG1和RAG2各由2个外显子组成,其编码区位于第2外显子中,分别编码1043和527氨基酸,位于人类染色体的11p13。RAG1蛋白的核心氨基酸区域为387~1 008,其编码产物Rag1蛋白形成的复合物是最重要的重组酶之一。该基因完全突变导致功能丧失,阻断V(D)J重组,导致成熟B细胞和T细胞功能完全缺失。复合杂合突变是两条同源染色体的相同基因座上有两个突变等位基因的杂合基因型,亚等位基因复合杂合突变允许低水平重组酶活性,突变RAG1基因重组酶的活性为野生型活性的5%~50%,保留了部分T和B细胞的发育。因此,RAG1完全突变和亚等位基因复合杂合突变可引起不同的临床表现和免疫表型。Lee等[5]利用流式细胞术,通过绿色荧光蛋白表达,检测不同RAG1基因突变引起的重组酶活性,证实了RAG1基因突变引起不同临床表型与重组酶活性存在关联。RAG1基因完全突变导致SCID[6,7,8],亚等位基因复合杂合突变导致OS[8,9,10,11,12]、泄露型SCID[3]和其他一系列临床表现,γδ+TCR T细胞的自体扩增[13]、自身免疫性疾病[8,13,14]、肉芽肿形成[9, 14]、肝脾肿大[8,11,12]、无丙种球蛋白血症[15]、普通变异型免疫缺陷病(common variable immunodeficiency,CVID)[16]和选择性IgA缺乏症[17]等。2017年,Schröder等[18]报道1例成人RAG1基因复合杂合突变引起进行性多灶性脑白质病。本研究中,5例纯合RAG1基因纯合突变均为已报道突变点位,例4基因突变位点为c.2459A>G,例2为c.2923G>A,其余3例基因突变位点均为c.2332C>T,临床表型为SCID和OS。例1、3、6为RAG1基因复合杂合突变,临床表型为泄露型SCID(例1、例3)和CID(例6)。例6行全外显子基因检测仅发现RAG1复合杂合突变,基因突变位点为c.2332G>A(父亲来源)和c.2103_2103delinsCAGAA(母亲来源),考虑为RAG1基因突变引起。结合年龄,4例IgM和IgG升高,可能为T或B细胞没有完全丧失数量或功能,可以分泌抗体,反复或持续感染引起。
5例RAG1基因纯合突变的临床表型为SCID和OS。OS患儿和1例SCID患儿转入血液科行造血干细胞移植,其余均已死亡。例5与例8为亲兄弟,当例5是确诊RAG1突变SCID时,医生已明确告知父母,如再次生育时需产前检查,但家属未予重视,导致再次不良后果的发生。SCID一般在生后6个月内起病,临床表现重,感染不易控制,易发生条件致病菌感染,且病情反复、迁延不愈,部分出现持续性腹泻。病原体包括细菌、真菌、病毒和(或)卡介苗等[19]。本研究中4例SCID均有呼吸道感染,3例有消化道感染,1例出现泌尿道感染、中枢神经系统感染,1例发现肺部、腹腔多处结节性占位伴多发骨质破坏。病原学阳性发现表皮葡萄球菌、嗜麦芽窄食单胞菌、人葡萄球菌、抗酸杆菌,还发现真菌和CMV感染,并出现持续轮状病毒感染。例2为OS,生后反复、严重皮疹,病初医生诊断为湿疹,经抗过敏及激素软膏等处理后效果不佳。随着病情发展,渐渐出现"红皮病"表现,无秃头症,并有轮状病毒引起的持续腹泻,肝脾淋巴结肿大,实验室检查外周血嗜酸性粒细胞和IgE增高。2015年,Matthews等[10]也报道了1例4月龄OS患儿确诊为RAG1基因复合杂合突变,分别来自母亲的无义突变和父亲的错义突变,免疫表型为T+B-,与例2的免疫表型不一样,可能与基因突变位点区域相关[12]。临床上很难区分是母体细胞植入所致的移植物抗宿主反应(graft-versus-host disease,GVHD)样皮疹与OS的严重湿疹。所有病例均未用短片段重复序列(STR)方法检测母体细胞植入情况,存在一定不足。2017年,我院开始进行母体细胞植入的研究,10例中6例发现母体细胞植入现象,均没有皮肤表现(数据未发表)。Wahlstrom等[20]报道47.3%的SCID出现母体细胞植入现象,但未发现OS病例。
3例RAG1基因复合杂合突变的临床表型为泄露型SCID和CID,男2例,女1例,高于纯合突变病例。1例也转入血液科行造血干细胞移植,1例出院门诊随访,1例死亡。3例SCID均有呼吸道感染,其中1例出现支气管扩张,2例有消化道感染,1例视网膜血管炎,病原学发现肺炎链球菌、大肠埃希菌、真菌和巨细胞病毒。Schuetz等[9]报道了2例泄露型SCID病例,表现为皮肤肉芽肿形成和反复肺部感染,病初予常规治疗效果不佳,分别在2岁和7岁时确诊RAG1基因复合杂合突变,行造血干细胞移植后症状消失。例1淋巴细胞亚群提示CD3+T淋巴细胞占76.69%,CD4+T淋巴细胞占7.1%,CD8+T淋巴细胞占19.75%,精细分型表现为双阴性T细胞明显升高。在功能方面,双阴性T细胞细胞具有抑制TCR的CD4+和CD8+ T细胞的功能,以及抑制性免疫调节作用[21]。例6在2月龄始即有反复呼吸道感染病史,当地医院给予丙种球蛋白(IVIG)等治疗,13岁就诊我科时提示IgM明显升高,已出现支气管扩张,建议如果患儿病情继续进展,可行造血干细胞移植治疗。2016年,Cifaldi等[15]也报道了1例男孩,6岁时被诊断为无丙种球蛋白血症,规律IVIG治疗,12岁确诊为RAG1基因复合杂合突变,但作者指出对于轻度和延迟出现症状的RAG缺陷患者,是否行造血干细胞移植治疗尚存争议。本研究主要不足是未对病例进行RAG1基因编码的重组酶活性进行检测,这样可更好证实复合杂合基因突变是否为临床病因,可作为今后研究的方向。
我国卡介苗、脊髓灰质炎和水痘疫苗等减毒活疫苗为国家计划免疫接种疫苗,其中卡介苗排除禁忌证后于生后24 h即接种。卡介苗病即接种卡介苗后发生的感染,包括淋巴结炎、肺、胸膜、脑膜等播散性感染时有发生,严重者可致死。由于存在T淋巴细胞缺陷,包括RAG1基因突变的SCID患儿不能接种此类活疫苗,可导致相应活疫苗接种感染。本研究中2例出现卡介苗同侧腋下淋巴结肿大,1例穿刺液涂片抗酸染色阳性,给予抗结核分枝杆菌治疗。李惠民等[22]报道18例播散性卡介苗感染中,PID12例,其中SCID有3例。这说明早期发现和排除免疫缺陷病非常重要,特别是家族有早期夭折或PID病史,婴儿出生后应暂停接种卡介苗等活疫苗,避免发生疫苗接种严重不良反应。
CMV感染是T淋巴细胞缺陷的标志之一,也是最常见的机会性感染。结合CMV-DNA滴度增高,本组诊断CMV感染5例,1例引起了视力损害,其中2例CMV-IgM阳性,说明感染后仍可能产生IgM抗体。确诊轮状病毒肠炎4例,复查大便轮状病毒抗原均阳性。病毒基因组可能是DNA或是RNA,通过与宿主表面的相应受体结合而进入机体,随后就在宿主或病毒蛋白酶的作用下开始病毒基因组的复制并合成病毒mRNA,均依赖于宿主细胞进行病毒蛋白的翻译和子代病毒颗粒的组装。CD8+T细胞对多数病毒的免疫防御至关重要,因为病原体在细胞内复制,病毒抗原被呈递在感染细胞表面的主要组织相容性复合体(MHC)Ⅰ类分子上,再与CD8+T细胞分子结合,引发一系列免疫反应,达到清除病毒的目的。对于感染CMV的SCID患儿,由于T细胞功能低下相关,不能有效清除体内感染的病毒,可能更易引起视力及听力等并发症。有研究报道CMV感染的SCID患儿,可刺激γδ+TCR T细胞的自体扩增[13]。2012年,Patel等[23]报道一例SCID患儿,2月龄时确诊轮状病毒感染,持续的病毒血症,并有慢性间歇性腹泻,直至移植成功后2个月(年龄11.5月龄),轮状病毒感染指标转阴性。
不同RAG1基因突变患儿的临床、免疫表型多样,起病早,确诊年龄可早或晚,易感病毒、细菌、真菌、结核等多种病原体。针对反复、严重感染,特殊或多病原、常规治疗效果欠佳的感染,慢性腹泻,生长发育落后和(或)有早期夭折家族史的患儿,临床医生应对患儿进行临床和实验室的免疫功能评价,发现异常尽早进行基因检测,及早诊断,及时修正治疗方案,如造血干细胞移植,以防止自身免疫性疾病、恶性肿瘤和致命性感染的发生。
参考文献(略)
欢迎分享到朋友圈,如需转载,请注明转载自《中华儿科杂志》
Strategy
﹀
﹀
﹀
或






