本文刊于:中华儿科杂志, 2018,56(4) : 247-260
作者:《中华儿科杂志》编辑委员会 中华医学会儿科学分会呼吸学组肺血管疾病协作组 中华医学会儿科学分会呼吸学组弥漫性肺实质/肺间质性疾病协作组
前言
儿童疾病谱中最具特色的是与发育相关的疾病。各器官或系统在胚胎期经历了信号通路网络的精准时空调控后,才能在出生后发挥正常的生理功能。这一发育的调控在胚胎孕育过程中可能受到多种因素的干预,如果这种影响因素来自遗传物质(染色体或基因),出生后形成的就是遗传代谢性疾病;如果影响来自外界或母体的非遗传因素,胎儿发育过程的停滞、变异则会造成出生后的各种畸形。这类在胚胎孕育过程中形成的疾病总称为出生缺陷(birth defects)或者先天性疾病,概括了遗传性和不良环境等原因引起的、出生时存在的各种结构性畸形和功能性异常。
从发生学上看,呼吸系统发育时间早且持续时间长。呼吸系统的先天性疾病种类繁杂,如何分类和统一命名,国内外文献尚无统一报道,至今仍存在一病数名和数病同名的现象。概念和分类是疾病研究的基础。随着近年来支气管镜技术和影像学技术的进步,对于先天性呼吸系统疾病的认识逐步深入。因此,有必要在概念厘清的基础上,从构架来规范这类疾病的分类。《中华儿科杂志》编辑委员会携手中华医学会儿科学分会呼吸学组的肺血管疾病协作组和肺间质性疾病协作组对国内外相关文献进行了查阅、回顾和分析,并经国内本领域的多位专家讨论、审核后提出了本分类建议,以期更好地认识该类疾病的发生发展过程,为临床诊治提供有益的帮助。
涉及呼吸系统的先天性疾病种类繁多。本分类建议仅列入以呼吸系统为原发缺陷部位和主要表征的先天性疾病,主要表征是指所有病例均存在的首发临床表现。本建议根据解剖部位和影像学特点,将儿童先天性呼吸系统疾病分为八类,分别是先天性上气道疾病、先天性下气道疾病、先天性肺泡及周围气道异常、先天性肺血管异常、先天性肺实质合并肺血管异常、先天性肺淋巴管疾病、先天性胸廓及膈发育异常、其他。中枢神经系统引起的呼吸系统异常不列入本建议。
【先天性肺泡及周围气道异常】
该组类型的解剖范围包括肺泡及17级细支气管以下的移行区气道,指各种原因所致的肺泡数量、体积、形态和功能的发育异常及移行区气道的异常(表3)。

重点介绍先天性肺表面物质代谢缺陷(inborn errors of surfactant metabolism,IESM)[21]。
一、肺表面物质蛋白(SP)B基因(SFTPB)变异(surfactant protein B gene variation)
曾用名称:SP-B缺陷(SP-B deficiency),肺表面物质代谢障碍1型(surfactant metabolism dysfunction, type 1)。
概述:由于SFTPB致病性变异造成肺表面物质功能丧失引起的弥漫性肺间质疾病,1993年报道的第一个IESM[22]。
病因:SFTPB位于第2号染色体,SP-B在降低肺泡表面张力、SP-C前体蛋白(proSP-C)的加工、肺表面物质磷脂在环层体中的排列和维持新生早期的肺功能起关键作用。目前可以识别有40多种SP-B基因的不同变异,本病属于常染色体隐性遗传。肺泡蛋白沉积症(PAP)是主要的病理类型。
分类和临床特点:SP-B完全缺陷的表现为足月新生儿出生时进行性呼吸窘迫,临床和影像学与早产儿严重的呼吸窘迫综合征(RDS)相一致。但对机械通气和肺表面物质替代治疗反应不佳,常在3~6月龄内死亡。少数SP-B部分缺陷的婴儿开始症状轻微,可能存活数月至数年。
诊断:依据临床和胸部X线或CT表现可怀疑本病,确诊需要基因检测。
二、SP-C基因(SFTPC)变异(surfactant protein C gene variation)
曾用名称:SP-C缺陷(SP-C deficiency),肺表面物质代谢障碍2型(surfactant metabolism dysfunction, type 2)。
概述:SFTPC致病性变异造成肺表面物质功能障碍,引起的弥漫性肺间质疾病。2001年首次报道[23]。
病因:SFTPC位于第8号染色体,主要作用是稳定磷脂,稳定表面物质。目前已知有50多种基因变异类型。本病属于常染色体显性遗传。病理学表现为婴儿慢性肺泡炎、非特异性间质性肺炎和脱屑性间质性肺炎等。
分类和临床特点:发病年龄和严重程度变异相当大,包括从新生儿严重RDS到婴儿、儿童,甚至成年人才出现症状。10%~15%在新生儿期出现呼吸道症状,可在新生儿期死亡;约40%在生后1~6个月出现症状,表现为弥漫性肺部疾病,包括呼吸急促、低氧血症和生长受限等。
诊断:对于临床符合且不明原因的弥漫性肺间质疾病应考虑本病,确诊需要基因检测。
三、ATP结合盒A转运家族成分3基因(ABCA3)变异(ATP-binding cassette family of transports member A3 gene variation)
曾用名称:ABCA3缺陷(ABCA3 deficiency),肺表面物质代谢障碍3型(surfactant metabolism dysfunction, type 3)。
概述:由于ABCA3致病性变异造成肺表面物质功能障碍引起的弥漫性肺间质疾病,2004年首次报道,为最常见IESM[24]。
病因:ABCA3位于16号染色体(16p13.3),功能是把脂类转运到环层体,与SP加工,维持表面物质中磷脂的稳定。目前已确定有150多种变异,为常染色体隐性遗传。病理可表现为PAP、脱屑性间质性肺炎、非特异性间质性肺炎和婴儿慢性肺泡炎。
分类和临床特点:ABCA3基因变异的临床表现和影像学表现与SP-B、SP-C变异重叠。在新生儿表现为致死性RDS,其影像学改变及临床症状与SP-B缺陷相同。在婴儿或年长儿表现与SP-C变异类似。ABCA3基因变异和SP-C缺陷同时存在时,病情更加严重。
诊断:对于临床符合且不明原因的弥漫性肺间质疾病应考虑本病,确诊需要基因检测。
四、粒-巨细胞集落刺激因子受体基因(GM-CSFR)变异(granulocyte-macrophage colony-stimulating factor receptor gene variation)
曾用名称:先天性(congenital pulmonary alveolar proteinosis)、遗传性肺PAP(hereditary pulmonary alveolar proteinosis)、肺表面物质代谢障碍4型(surfactant metabolism dysfunction, type 4)。
概述:GM-CSFR致病性变异或缺失,造成肺巨噬细胞不能有效清除SP而引起的PAP。2008年首先证实其为家族性PAP的致病原因[25]。
病因:GM-CSFR的α链或β链基因变异或缺失,引起肺巨噬细胞接受GM-CSF信号出现障碍,进而肺巨噬细胞不能成熟,无法分解和清除SP。属于常染色体隐性遗传。
分类和临床特点:多数在婴幼儿及儿童发病,表现为弥漫性肺间质表现如呼吸增快或运动不耐受、呼吸困难、生长落后、呼吸衰竭等,胸部影像学及肺泡灌洗液或肺活检符合PAP。先天性PAP也见于SP-B、ABCA3基因缺陷。
诊断:对于临床、影像及病理符合PAP的患儿,在除外继发原因以后应考虑本病,确诊需要基因检测。
五、甲状腺转录因子-1基因(NKX2.1)变异(thyroid tranion factor 1 gene vanation)。
曾用名称:肺-脑-甲状腺综合征(lung-brain-thyroid syndrome)。
概述:NKX2.1致病性变异,造成甲状腺、脑和肺发育障碍。2000年首次报道了先天性甲状腺功能低下合并反复呼吸窘迫的患儿[26]。
病因:NKX2.1位于16号染色体(14q13.3),其编码的甲状腺转录因子-1(TTF-1)在甲状腺、脑和肺表达,在多种基因的表达中发挥转录调控作用,包括SFTPB、SFTPC和ABCA3。其变异可以影响肺、甲状腺和脑的形态和功能发育,也影响肺表面活性物质的代谢。为常染色体显性遗传,也可散发出现。
分类和临床特点:肺部病变可以从新生儿致死性RDS到儿童期慢性肺病、反复肺部感染。常伴甲状腺功能低下和神经症状(肌张力低下和舞蹈症)。但各个器官表现的程度可以不同,或仅有一个器官的表现如甲状腺功能低下、肌张力低下和舞蹈症等。
诊断:有肺、脑、甲状腺受累的表现高度可疑,确诊需要基因检测。
【先天性肺血管异常】
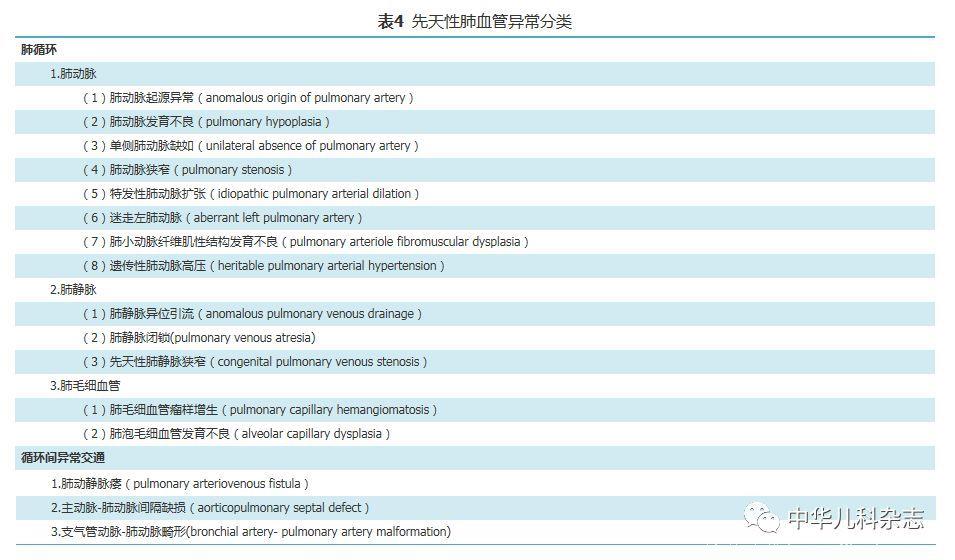
肺脏是体内唯一拥有两套血液循环系统的器官。其中,肺循环主要功能是气体的运输和交换,是肺的功能血管网;体循环的支气管动、静脉主要供应支气管、肺组织的血供,是肺的营养性血管。两套血管通过吻合口支相互交通。常见的先天性肺血管异常见表4。本分类中未包括体循环中的大血管异常以及合并了心脏畸形的肺血管异常。
一、遗传性肺动脉高压(heritable pulmonary arterial hypertension)
曾用名称:家族性肺动脉高压(familial pulmonary arterial hypertension)。
概述:特指限于当时技术条件找不到病因但有家族发病倾向或明确的已知基因异常的一类原发性肺动脉高压。这一概念的前身是家族性肺动脉高压,在2008年美国橘子郡举行的第4次全球肺高压专题会议上首次提出。
病因:目前已知病变有包括BMPR2、ALK-1、ENG、SMAD9、CAVl、KCNK3等6个基因的变异[27]。
分类和临床特点:根据2013年尼斯举行的第6次全球肺高压专题会议更新的分类标准,遗传性肺动脉高压属于肺高血压分类中的第一类"肺动脉高压"中的1.2亚类。可有劳累性呼吸困难、晕厥、胸痛、乏力、紫绀等表现。遗传性肺动脉高压患儿因肺循环压力增高,会导致毛细血管前肺小动脉及各级分支或肺泡毛细血管破裂出血,在病程中晚期合并咯血。
诊断:超声心动图、心电图、胸部影像学有助诊断,心导管检查是诊断金标准。儿童肺动脉高压的血流动力学定义建议采用如下标准:海平面状态下静息时,右心导管检查平均肺动脉压力≥25 mmHg(1 mmHg=0.133 kPa);肺动脉楔压≤15 mmHg[28]。在这一基础上,如果基因检测存在上述6种基因之一的致病性突变,或者有明显家族发病倾向,可诊断本病。
二、迷走左肺动脉(aberrant left pulmonary artery)
本病属于"血管环"的一种。"血管环"畸形是一组先天性疾病的总称,由于胚胎期多对鳃弓和成对的背侧主动脉未能顺序融合和吸收,主动脉和肺动脉发育畸形可在解剖位置上形成围绕在气管和食管周围的"血管环",引起呼吸道和(或)消化道压迫,临床上主要表现为喘息和吞咽困难[29]。1737年有学者描述了双主动脉弓,1939年有了血管环的概念,1945年学者们对"血管环"畸形进行了分类,包括3大类:(1)主动脉弓异常,包括双主动脉弓、右位主动脉弓合并左侧肺动脉韧带、迷走右锁骨下动脉以及较罕见的颈位主动脉弓;(2)迷走左肺动脉(左肺动脉起源异常或肺动脉吊带);(3)迷走无名动脉(无名动脉压迫综合征或头臂干压迫综合征)。这6种畸形涉及了肺循环或体循环,因此表4中仅列出涉及肺循环的一种畸形"迷走左肺动脉"。
曾用名称:肺动脉吊带(pulmonary artery sling,PAS)、左肺动脉异常起源于右肺动脉。
概述:指左肺动脉异常起源自右肺动脉,走行在食管与气管之间,并环绕右主支气管和气管远段,到达左侧肺门,在气管远端和右主支气管近端形成吊带,相邻气道和食管有不同程度受压。1897年首次报道,1958年获命名。
病因:起源于左侧主动脉第六弓的左肺动脉发育不良,右侧第六弓取代形成。
分类和临床特点:肺动脉的异常起源包括左肺动脉或左上肺动脉两类[14]。反复喘鸣、阵发性呼吸困难和反复的肺部感染是患儿就诊的主要原因。部分患儿伴气管、支气管狭窄及完整的气管软骨环,多合并气管软化和(或)心内畸形。
诊断:心血管造影、MRI和CTA均有助于肺动脉吊带的诊断[30]。CTA还能显示左肺动脉或左下肺动脉与气管和食管的关系。
三、先天性肺静脉狭窄(congenital pulmonary venous stenosis)
概述:指各种因素导致的肺静脉管腔狭窄,导致肺静脉血液回流受阻的先天性疾病。1951年首次报道。
病因:尚不清楚。病理表现为非特异性的内膜纤维化、纤维增厚导致肺静脉管腔狭窄,可1条或多条受累。
分类及临床特点:狭窄的部位多见于肺静脉与左房连接处,可分为管状狭窄和隔膜型狭窄。症状取决于静脉受累的数目、狭窄程度及合并畸形的情况。患儿可有反复呼吸道感染、运动耐量下降,个别在剧烈活动时可出现晕厥,最终发生右心衰竭。其他非特异症状包括发育延迟、反复咯血和中度以上肺动脉高压。
诊断:超声心动图是首选的无创检查手段,而血管造影的诊断价值最大,MRI和CT血管造影(CTA)也具有诊断意义[31]。
四、肺毛细血管瘤样增生(pulmonary capillary hemangiomatosis)
概述:是一种罕见的肺组织内血管增殖性疾病,预后不良。大量薄壁毛细血管增生并浸润肺泡壁、气道甚至胸膜,导致低氧血症及肺动脉高压。1978年对肺毛细血管瘤样增生进行了首次描述。
病因:不明,有报道部分病例中检出EIF2AK4基因变异[32]。
分类和临床特点:根据2013年尼斯会议肺高压分类标准,本病属于第一类肺动脉高压中的1'亚类。逐渐加重的活动后气短是最常见的首发症状,随病情进展,右心功能衰竭的表现将更为明显。其他症状包括咳嗽、咯血、乏力、胸痛等。
诊断:准确的诊断需要综合临床、影像及组织病理三个方面的资料,其中病理组织学检查被公认为是最可靠的手段,对确诊具有决定性作用[33]。肺毛细血管瘤样增生的特征性组织学改变为肺泡壁毛细血管增生,早期表现为多排毛细血管沿肺泡壁分布;晚期毛细血管呈结节或片状排列,管腔不明显。病情进展中可见内膜纤维化和继发性静脉阻塞。
五、肺动静脉瘘(pulmonary arteriovenous fistula)
曾用名:肺动静脉畸形(pulmonary arteriovenous malformations)。
概述:由于肺动脉与肺静脉之间出现异常交通,肺动脉血未经肺泡氧合直接流入肺静脉形成右向左分流导致低氧血症的畸形,常伴有遗传性出血性毛细血管扩张症[34]。1897年首次报道。1940年首次外科治疗成功。
病因:由于胚胎发育时肺动脉支与静脉丛间的血管间隔形成发生障碍,毛细血管发育不全或退化,肺动静脉之间直接形成交通。目前认为endoglin (ENG)、activin receptor-like kinase 1 (ALK1)和Sma and madmomologue (SMAD4)基因变异可能与本病有关。
分类和临床特点:病理学分两型:Ⅰ型,囊状肺动静脉瘘,包括单纯型和复杂型,单纯型囊状肺动静脉瘘最常见,输入和输出血管各一支,交通血管呈瘤样扩张,病灶可单发或多发;复杂型囊状肺动静脉瘘输入和输出血管各多支,交通血管瘤囊常有分隔。Ⅱ型,弥漫型肺小动静脉瘘,少见,呈两肺散在多发的微小动静脉瘘。临床症状与瘘的多少及分流量大小有关,分流量少的早期可无症状;分流量大者,幼儿或学龄前出现症状。最常见的症状为气促或呼吸困难,病初为活动后呼吸困难,随着年龄增长和瘘管的扩张,静息时也可发生呼吸困难,可有杵状指(趾)。
诊断:胸部X线是筛查肺动静脉痿的常用检查之一,CTA是诊断肺动静脉痿的首选无创检查手段,具有较高的敏感性,DSA是诊断肺动静脉痿的金标准。
【先天性肺实质合并肺血管畸形】
本组疾病包括两个临床常见的类型:肺隔离症和弯刀综合征。
一、先天性肺隔离症(congenital pulmonary sequestration)
曾用名称:支气管肺隔离症(bronchopulmonary sequestration),肺隔离症(pulmonary sequestration)。
概述:指存在于胸腔的由体循环供血的胚胎肺组织,该组织与支气管树和肺血管无连接,也没有正常的呼吸功能。最早报道见诸于1777年。1946年首次使用"肺隔离症"概念。
病因:多为假腺管期肺组织部分发育异常所致,有副肺芽等多种假说。
分类和临床特点:分为叶外型和叶内型两种类型。叶内型由肺静脉供血,局部易反复发生感染,表现为咳嗽、咳痰、发热、咯血、胸痛等,当病灶较大压迫邻近正常肺组织时,则出现胸闷、气短。可能出现局灶性小血管破裂出血引起咯血。叶外型由体静脉供血,多无明显症状。但合并畸形者多见。
诊断:胎儿时期超声检查和MRI具有重要价值。出生后胸部X线片、超声、CT、MRI、血管造影均具有诊断价值[35]。
二、弯刀综合征(scimitar syndrome)
曾用名称:军刀综合征、HALASZ综合征、镜像肺综合征、肺发育不良综合征(hypogenetic lung syndrome)、支气管-右肺动脉综合征、腔静脉-支气管血管综合征。
概述:部分性肺静脉畸形引流的一种类型,其特点为右肺静脉引流开口于下腔静脉,少见引流至门静脉、肝静脉或右心房。因异常、弯曲的引流入下腔静脉的右肺静脉在胸部X线片显示类似弯曲的刀影,故名"弯刀综合征"。1960年首次报道。临床罕见。
病因:不明,多数学者认为与胚胎期肺的发育异常有关。
分类和临床特点:可分为婴儿型、儿童或成人型两种类型[36]。婴儿型的弯刀综合征通常伴严重的呼吸窘迫、肺动脉高压、反复肺部感染、发育迟缓和心力衰竭,常合并其他先天性心脏病和(或)肺动脉高压,临床症状较重,容易出现呼吸困难、反复肺部感染及充血性心力衰竭、生长迟缓等。半数患儿被诊断时并无症状或症状很轻。
诊断:胎儿时期超声检查或MRI具有重要价值。出生后胸部X线片、超声、CT、MRI、血管造影等均具有诊断价值。
参考文献(略)
欢迎分享到朋友圈,如需转载,请注明转载自《中华儿科杂志》
Strategy
﹀
﹀
﹀
或




-mw.jpeg)
